

News & Insights
Capture the beautiful moments and wonderful memories
【Literature Express】Modifying the host range of phage and inhibiting bacterial resistance through phage tail fiber mutation
Release time:
2025-03-04
To address this issue, the author's research objective is to engineer bacteriophages, modifying key regions of the phage tail fiber to expand their host range and prevent the development of resistance.

Research Overview
Kevin Yehl et al. published a paper titled "Engineering phage host range and suppressing bacterial resistance through phage tail fiber mutations" in Cell. In the article, the authors identified the host range determining region (HRDR) in the T3 phage tail fiber protein through natural evolution and structural modeling, and developed a high-throughput strategy to genetically engineer these regions through site-directed mutagenesis, generating synthetic phages. The mutated HRDR produced phages with altered host ranges, and the selected phages were able to suppress bacterial growth in vitro for a long time by preventing the emergence of resistance, and worked in vivo using a mouse model. This method is expected to help create next-generation antimicrobials that slow the development of resistance, and may be extended to other viral scaffolds for broader applications.
Research Background and Objectives
The exacerbation of drug-resistant bacterial infections and the scarcity of antibiotic resources have necessitated the development of new alternative therapies. Phage therapy has received widespread attention due to its different mechanism from antibiotics, which can bypass resistance issues. However, phages rely on host cell receptors to recognize and infect bacteria, and mutations in receptors can lead to phage resistance. Therefore, phage-based therapies typically consist of multiple phages to target different receptors, reducing the emergence of resistance.
To address this issue, the authors' research objective was to engineer phages, modifying key regions of the phage tail fibers, to expand their host range and prevent the development of resistance. By targeting these regions closely related to host recognition, increasing phage diversity, thereby improving phage adaptability and effectiveness.
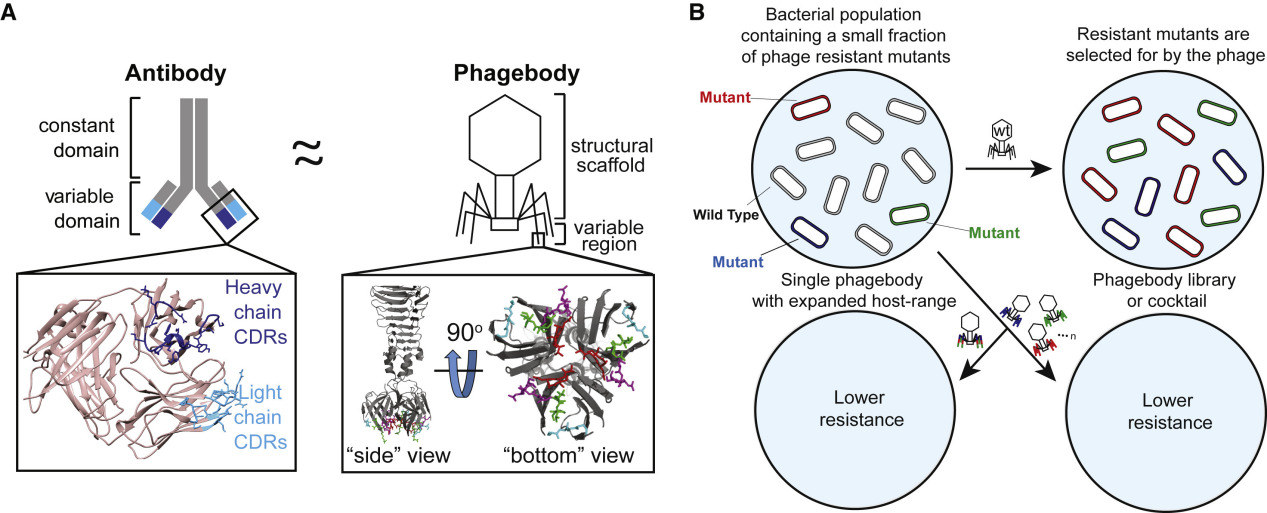
Figure 1. Phage design and its potential ability to target bacterial mutants
Research Content and Results
1. Identification of the Host Range Determining Region of T3
T3 initially binds to bacterial LPS via its tail fiber for host recognition. Each tail fiber consists of a homotrimer of the gene 17 product gp17, and the authors have previously identified the carboxyl terminus, the 450-553 amino acid globular domain, or the "tip" of gp 17 as a determinant of host specificity. The structure of the tip region (residues 454-558) of T3 gp17 was used using the published T7 gp17 structure (Figure 2). This structure was used to identify potential host-determining regions of T3 (Figure 2B). The distal 104 aa portion of gp17 forms an intertwined globular domain, whose shape is formed by an eight-stranded β-barrel (labeled strands B to I). These strands are connected by irregular loops, represented by the β-strands they connect. Three of these loops, CD, EF, and GH, face towards the tail fiber axis (inward loops) and are hypothesized not to be involved in host recognition. The other four loops, BC, DE, FG, and HI, are exposed on the opposite side of the β-barrel and point away from the tail fiber (Figure 2A and 2B; outward loops). It is hypothesized that these loops may interact with the host surface and may be the host range determining regions (HRDRs). 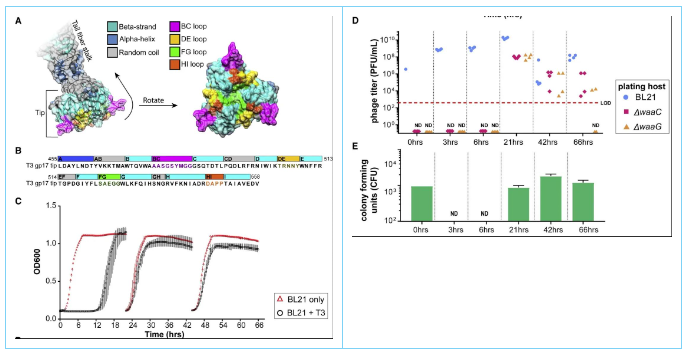
Figure 2. Computational structure of T3 gp17 and the emergence of resistance to T3 infection
Natural phage mutants were selected by infecting BL21 with T3 for 3 days, re-inoculating into fresh medium daily (Figure 2C). The generation of phage mutants is because T3 eliminated wild-type (WT) bacteria and selected resistant bacterial mutants, which in turn selected T3 mutants. Based on sequencing and complementation assays, most bacterial mutants contained mutations that altered the main receptor for T3 in the LPS biosynthesis pathway. Since LPS mutations are the major evolutionary pathway for bacteria to develop resistance to T3 infection, two LPS mutants of BL21 were constructed: the BL21 ΔwaaG mutant lacks the outer core of LPS, including the glucose moiety that serves as the receptor for WT T3, and the BL21 ΔwaaC mutant has almost no LPS (Figure 3B). Both strains contained truncated LPS and were tested for phage mutants with expanded host range (growth at 24, 48 and 72 hours). Phage mutants could be detected after 24 hours of co-culture (Figure 2D, red and orange data points), while bacterial resistance emerged 12 hours after infection (Figure 2C and 2E). 34 plaques were randomly isolated from BL21 ΔwaaG and ΔwaaC mutants and submitted for Sanger sequencing of gene 17. Of the 34 isolated phages, only 10 contained unique mutation combinations in gene 17, consisting of one mutation in the BC loop, one mutation in the FG loop, and four mutations in the HI loop. Surprisingly, although a large proportion of phage mutants could infect BL21 LPS mutants, the lysates failed to suppress bacterial resistance in WT BL21 (Figure 2C, Figure 2E). In summary, this suggests that phage evolution is too constrained to suppress resistance.
2. Strategy for introducing genetic variation into the HRDR
Since each of these loops is relatively small (4-9 aa long), a large diversity is generated by replacing each codon in the target loop with an NNK codon (Figure 3A), so that the loop sequence is completely randomized at the DNA level. To construct this library, the tail fiber gene 17 was cloned into a plasmid and PCR amplified using degenerate oligonucleotides designed to replace individual loops with a variable number of NNK codons. The plasmid library containing the mutated version of gene 17 was transformed into E. coli NEB5α and recombined into the T3 genome by infecting the transformants in early log phase. Infections were not carried out for more than 3 hours to minimize the enrichment of spontaneous phage mutants caused by random mutations acquired by DNA polymerase errors (Figure 2, C and 2E).
Because the entire sequence space of the BC loop could not be exhaustively sampled, three independent libraries were designed with partial randomization of the BC loop, where only the first four codons (BC[1-4]), the central five codons (BC[3-7]), or the last four codons (BC[6-9]) were random. The numbers in parentheses indicate the position of the codons within the loop (Figure S2A and S2B). In addition, compared to WT T3, the generated library contains elongated HI loops with one extra codon (HI[+1]) or three extra codons (HI[+3]) (Figure 3A).
3. Characterization of phage library and screening of bacterial LPS mutants
To quantify library diversity and determine potential library construction biases, high-throughput sequencing (NGS) (HiSeq) was performed at each step of library synthesis. To validate the hypothesis that loop randomization generates functional diversity, the phage library was screened on ΔwaaG and ΔwaaC LPS mutants (Figure 3B), both of which are T3-resistant strains. Each phage library was serially diluted and plated on these LPS mutant strains to quantify the number of plaque-forming units (PFU) and score the success of each library (Figures 3C and 3D). Screening the phage library on ΔwaaG and ΔwaaC bacterial mutants showed that mutagenesis of specific loops was more likely to expand the phage host range than others, highlighting key phage-host interactions. Each mutagenized HI loop library yielded phages effective against both ΔwaaG and ΔwaaC LPS mutants (Figure 3C). In contrast, phages from DE and FG loop mutagenesis rarely showed infectivity against ΔwaaG or ΔwaaC LPS mutants (Figure 3C). Approximately half of the libraries targeting all or part of the BC loop yielded phages capable of plaquing on ΔwaaG or ΔwaaC (Figure 3C). Therefore, it was concluded that mutagenesis of the HI and BC loops can generate productive phage mutants, while the DE and FG loops are sequentially constrained by tail fiber structure or do not play a role in receptor binding.
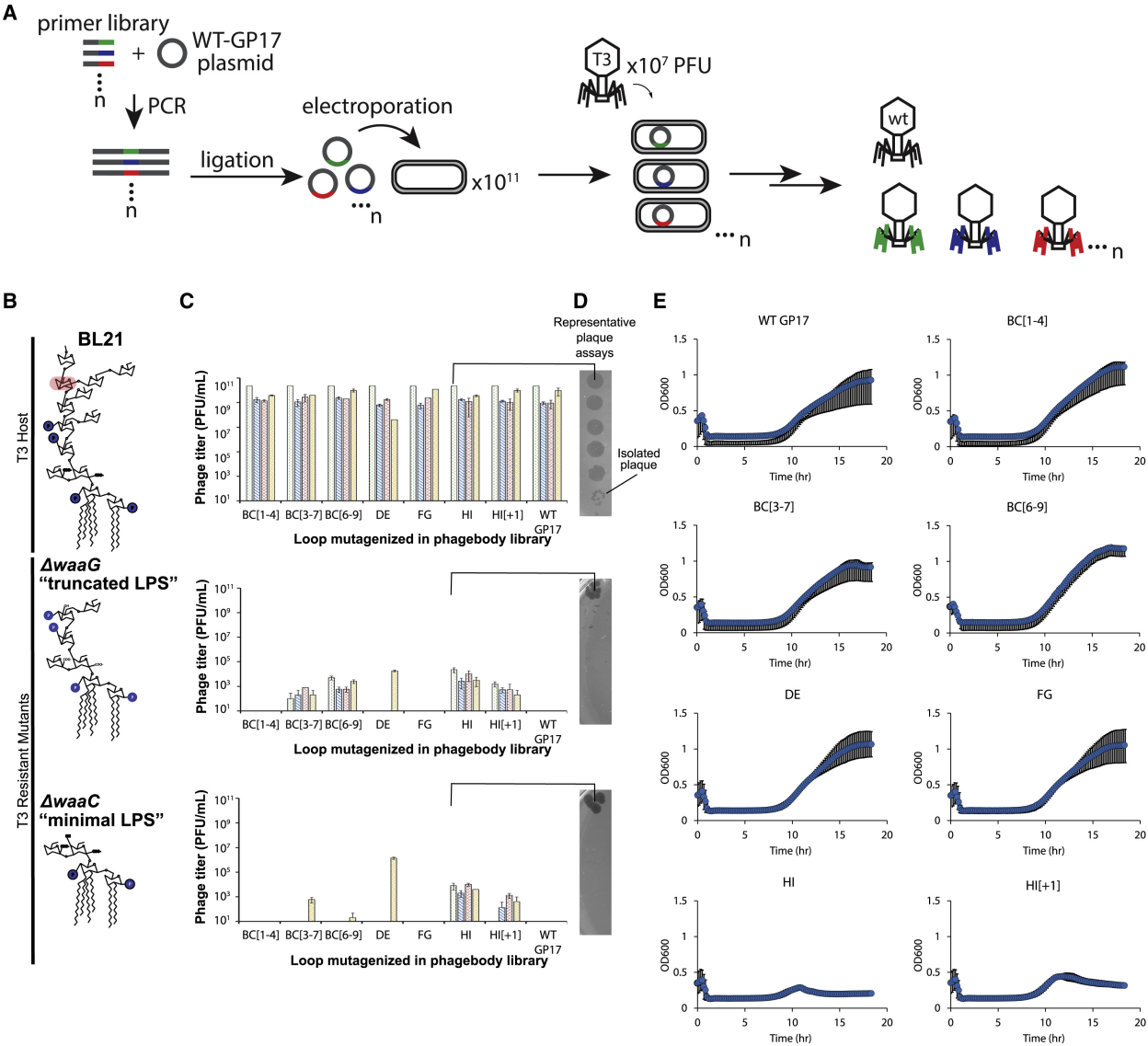
Figure 3 Phages show expanded host range against wild-type T3-resistant and LPS mutants that can suppress bacterial resistance in culture
4. Phages can delay or prevent bacterial resistance through loop mutations
Because phages can plaque and infect LPS mutants evolved from T3 infection, it was tested whether these phage libraries could reduce resistance compared to WT T3. Bacterial growth kinetics after phage infection were measured, and only the HI-targeted library prevented bacterial resistance 24 hours post-infection (Figure 3E). Surprisingly, the BC loop library did not reduce resistance despite the presence of LPS mutant-targeting phages. Some phages may lack function or at least fail to recognize their selected hosts. To mitigate this limitation, it was tested whether successive panning and amplification would uncover rare or poorly growing phages. To achieve this, three different mutant host strains were used, ΔwaaG, ΔwaaC, and from a T3-resistant BL21 mutant, called PRM01 (phage-resistant mutant). Panning experiments involved infecting fresh cultures of the desired bacterial strain with the phage library at a high multiplicity of infection (MOI = 0.4), then harvesting progeny phages and repeating the cycle 3 times at a lower MOI (Figure 4A). BC[3–7] and DE libraries still failed to yield phages capable of infecting any of the three tested strains. Phages from the FG library required an initial MOI = 40 (Figure 4B). FG libraries were also difficult to construct, as their transformation efficiency was consistently lower than other libraries, and diversity was lower. The remaining phage libraries readily yielded high titers of phages capable of infecting T3-resistant strains with only limited or no enrichment (Figure 4B).
Next, 10 individual isolated phages from the experiment were selected and mixed together in equal amounts to form a cocktail reagent. Four phages used in the mixture had mutations in the BC loop, one in the FG loop, and the remaining five in the HI loop. The cocktail-treated samples showed a decrease in bacterial titer over time (Figure 4C, orange squares), while the T3-infected cultures rebounded to saturation after 12 hours and maintained a constant bacterial concentration (Figure 4C, blue circles). The performance of the cocktail-treated cultures was significantly better than that of the T3-treated cultures (Figure 4C, blue circles vs. orange squares). By day 6, a large difference in bacterial load was observed between the mixture-treated and T3-treated cultures, with the phage mixture-treated samples showing a 5-log reduction in bacterial titer compared to T3 (Figure 4C, blue circles vs. orange squares). As a result, the identified phage mixture exhibited a similar ability to the HI library, despite having much lower genetic diversity within the phage population, and was able to prevent the regeneration of bacterial cultures in approximately one week. In contrast, T3 produced visible resistance after 12 hours and never managed to produce an effective response to control the bacterial population.
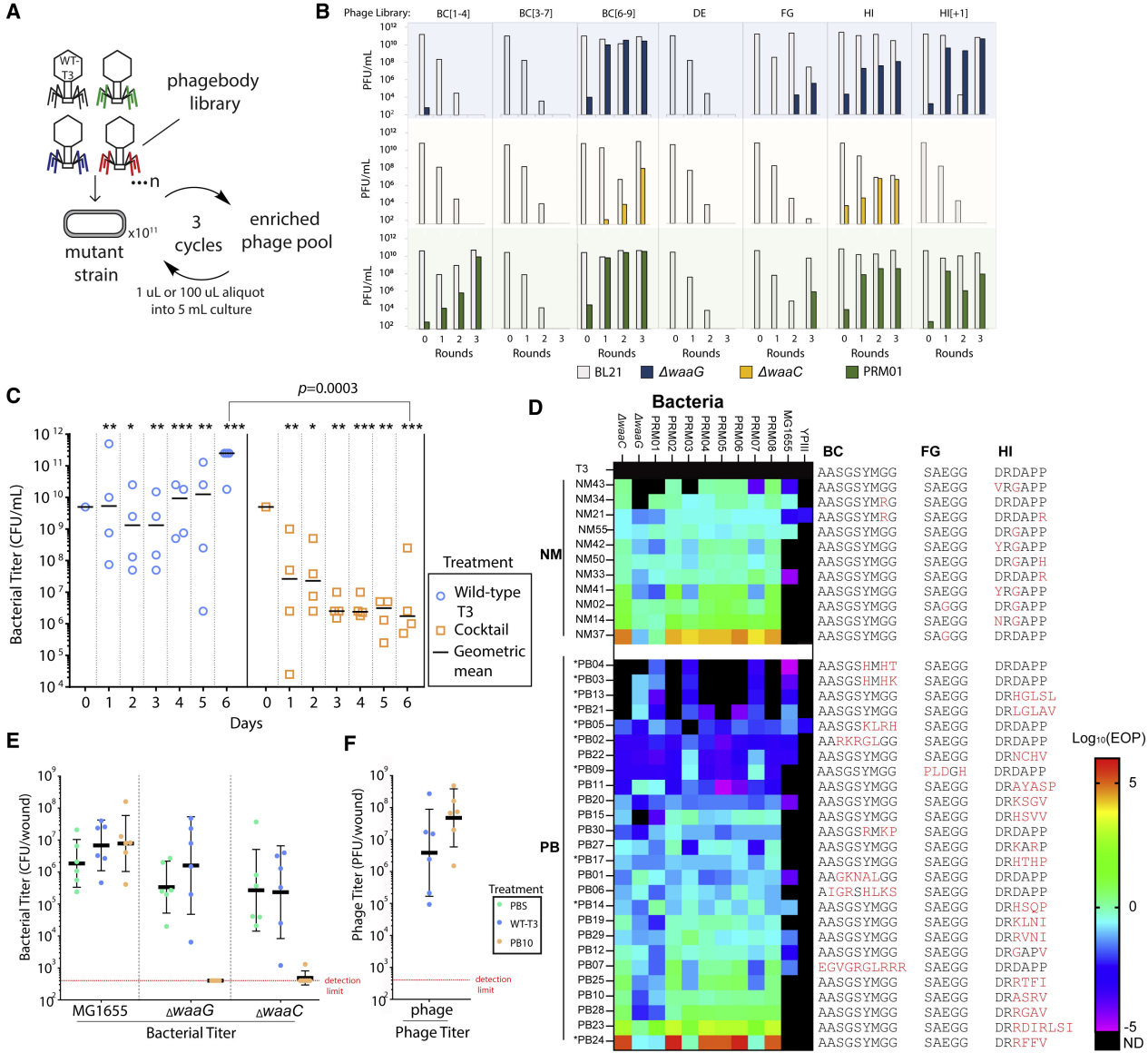
Figure 4 In vitro and in vivo activity of selected phages
5. Comparison between naturally evolved phages and phages with engineered HRDR
To better understand how phages are able to suppress bacterial resistance and whether there is a dependence on specific mutations or loop characteristics, additional natural mutants of phage (PB) and T3 (NM) were isolated to compare their genomes, host range, and killing efficiency. The host range was determined by plating PB and NM on a panel of BL21 mutants, Escherichia coli MG1655 (MG1655), and Yersinia pseudotuberculosis YPIII (YPIII). The latter two strains were included to determine the extent to which phage engineering expanded the host range, as T3 is known to be able to evolve infectivity for these two strains. BL21 included the initial three isolates (ΔwaaG, ΔwaaC, and PRM01) and seven additional randomly isolated BL21 mutants that acquired T3 resistance (PRM02-PRM08) (Figure 4D). Figure 4D summarizes the killing efficiency (EOP) of PB and NM, with the titer on BL21 as a reference. As shown in Figure 4D, most NMs showed a broad host range against BL21, with 8/11 NMs able to infect all 10 strains with efficiency similar to that of infecting WT BL21. The EOP ≥2 for most T3-resistant strains except ΔwaaG and PRM001. NM02 has the same mutation as NM37 (FG: E525G) but also contains an additional mutation (HI: D547G), which restores plaque ability on WT BL21. 5/11 NMs were able to infect MG16555, with only NM21 able to infect YPIII.
6. PB10 retains specificity and is active in a mouse skin infection model
To test whether the engineered phages are functional in vivo and maintain selectivity, phage PB10 was tested using a mouse skin infection model because it has broad infectivity against PRM and a hydrophilic HI loop; therefore, the authors anticipated that it would prevent the development of resistance. Anesthetized mice were scratch-infected and infected with an equal amount of a mixture of MG1655, ΔwaaC, and ΔwaaG. After 1 h of infection, they were treated with PBS, T3, or PB10.
The concentration of MG1655 in infected wounds did not change significantly due to the applied treatment (Figure 4E). Mice treated with PBS or T3 showed no significant reduction in ΔwaaG or ΔwaaC counts. On the other hand, in mice treated only with PB10, 6/6 showed no countable ΔwaaG colonies (Figure 4E), and 5/6 showed no countable ΔwaaC colonies (Figure 4E); one mouse had a very low ΔwaaC count. It is concluded that PB10 is able to replicate and clear ΔwaaG and ΔwaaC in wounds. Importantly, these results indicate that PB maintains selectivity and activity in animal tissues.
Discussion
This article presents a powerful and simple method for producing and screening engineered phage libraries with expanded host ranges, and demonstrates that these engineered phages can suppress bacterial resistance to phage infection. The method described focuses on generating viable phages with subtle host range alterations to target resistant mutants. Mutagenesis is directed to the expected highly efficient tail fiber regions, i.e., HRDRs. Because these regions are short, the potential sequence space can be saturated, producing large phage libraries with diverse functionalities ranging from more specific to broader host ranges. It is envisioned that high-throughput mutagenesis and characterization of host range determinants will become an important and useful tool for deciphering viral host recognition. As more viral tail components are structurally resolved and sequenced, the breadth of viral models to which this strategy can be applied will also expand.
Implications
This article provides innovative strategies in host range expansion, high-throughput screening, and in vivo validation, providing important technical references for our company's future R&D work.
Next article
Related News
Global Exclusive Agent: Handee Green Biotech Co.,Ltd




